O metabolismo da bilirrubina pode ser subdividido em captação, armazenamento, conjugação e secreção hepática, na qual se encontram enzimas cujas atividades podem ser alteradas causando processos patogénicos.
As hemácias são formadas na medula óssea e se destinam ao sistema circulatório, circulam por 120 dias e são destruídas. No transcorrer desse período, seu sistema metabólico torna-se cada vez menos ativo, a sua membrana fica mais frágil (senescente) e rompe-se durante sua passagem em lugares estreitos.
Muitas hemácias se autodestroem no baço, onde os espaços entre as trabéculas estruturais da polpa vermelha pelos quais devem passar a maioria das hemácias medem apenas 3 µm de largura em comparação com o diâmetro de 8 µm das hemácias.
A ruptura das hemácias libera a hemoglobina, que é fagocitada de imediato pelos macrófagos em muitas partes do organismo, especialmente pelas células de Kupffer, no fígado, e pelos macrófagos no baço e na medula óssea.
É captada pelo sistema retículo-endotelial, sendo transformada a sua hemoglobina pela hemeoxigenase em biliverdina, monóxido de carbono e ferro. A biliverdina-redutase converte a biliverdina em bilirrubina livre, sendo gradualmente liberada dos macrófagos para o plasma.
A taxa de conversão da biliverdina em bilirrubina livre é de 4 mg/kg/dia. Essa bilirrubina é lipossolúvel e apolar, podendo ligar-se à albumina e sua fração livre atravessar a barreira hematoencefálica.
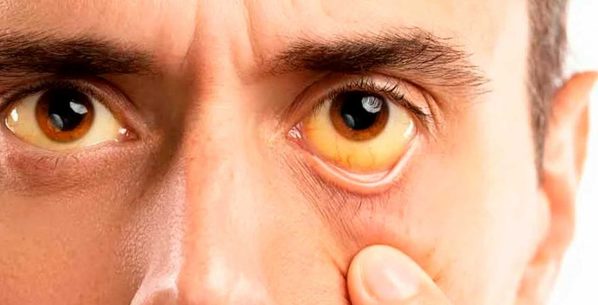
A bilirrubina livre atravessa facilmente a barreira hematoencefálica, sendo potencialmente tóxica para o tecido nervoso.
A afinidade da bilirrubina pelo tecido nervoso não conjugada concomitante às concentrações elevadas no sangue em recém-nascidos pode impregnar os gânglios da base, causando kernicterus.
A bilirrubina sérica se liga fortemente à albumina plasmática, sendo transportada por todo o sangue e fluido intersticiais.
Observa-se que a pequena fração de bilirrubina não ligada ao plasma pode aumentar na doença hemolítica grave ou quando drogas ligadoras de proteínas deslocam a bilirrubina da albumina.
A bilirrubina ligada às proteínas plasmáticas é denominada “livre”, a fim de distinguir-se da forma conjugada.
A bilirrubina livre, quando chega ao fígado, é recolhida pelos hepatócitos por meio de sistemas proteicos, transportadores de membrana (proteínas X e Y), num processo chamado captação.
É a seguir liberada da albumina plasmática e conjugada por ação de enzimas microssomais (UDP glicuroniltransferase) com uma ou duas moléculas de ácido glicurônico, formando um composto mais polar e hidrossolúvel, a “bilirrubina conjugada”.
Parte dessa bilirrubina liberada nos hepatócitos pode-se ligar a uma proteína citoplasmática denominada ligandina, etapa posterior à sua conjugação, que impede o efluxo dessa substância do hepatócito para o plasma.
A bilirrubina conjugada é excretada através do polo biliar dos hepatócitos que está em íntimo contato com os canalículos biliares e daí para os intestinos.
A conjugação da bilirrubina ocorre majoritariamente no fígado, sendo também observada nas células dos túbulos renais e nos enterócitos.
A bilirrubina conjugada é transportada como complexo lipídico-micelar até o duodeno através do ducto biliar principal, sendo desconjugada e reduzida no cólon por ação das glicuronidases bacterianas, formando os urobilinogênios (Figura 1).
Essas moléculas são excretadas nas fezes, em sua maioria, e pequena parte é reabsorvida através da mucosa intestinal e volta ao fígado pelo sistema porta, constituindo o ciclo entero-hepático da bilirrubina; e cerca de 5% são excretados na urina pelos rins.
A ligação forte da bilirrubina não conjugada com a albumina plasmática e o estabelecimento de interações fracas com os sais biliares, micelas mistas e vesículas lipídicas tornam-se sua excreção renal limitada, razão pela qual é eliminada, sobretudo pelo fígado.
Fonte:
Revista Médica de Minas Gerais
Bilirubin synthesis and metabolism, and hyperbilirubinemia associated with Gilbert’s Syndrome: A review of the literature